Our Research & Pipeline
There are 10,000 known rare diseases. Together, these diseases affect 300 million people globally, and 25 to 30 million people in the U.S. – or one in 10 Americans.
Despite great progress in recent years, 95% of rare diseases still do not have any approved treatments. That’s why Amicus remains so committed to our mission to develop and deliver transformative medicines for rare diseases.
Our belief in the fight to be at the forefront of therapies for rare diseases, like all of our work, is inspired by patients. From our earliest days through to today, we have welcomed patients into all aspects of Amicus, including our research. Their insights guide us to push the frontiers of science to bring forward breakthroughs for rare diseases as quickly as we can.
We’re incredibly proud to have brought forward two treatments for rare diseases for patients in our first two decades, and we’re fully committed to do more.
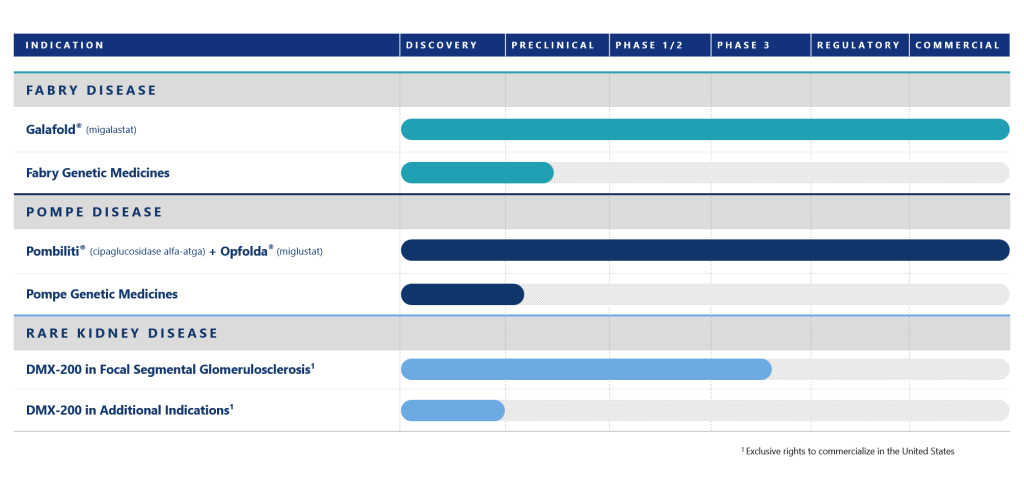
We continue to invest in R&D to obsolete our own technologies in Fabry and Pompe diseases.
In addition to this work, we are building a pipeline to bring hope to more patients with rare diseases. Each research program we pursue must:
In 2025, we expanded our U.S. pipeline with an investigational oral medicine for the rare kidney disease Focal Segmental Glomerulosclerosis.
Learn more about our pipeline here:
Pompe Disease is a rare lysosomal disease that causes muscle weakness. People with Pompe Disease are deficient in a certain type of enzyme, called acid alpha-glucosidase (GAA). The GAA enzyme is responsible for breaking down glycogen into glucose. When excess glycogen builds up in the lysosomes, it causes damage to the muscles throughout the body. This leads to mobility problems and respiratory issues as the muscles responsible for movement and breathing begin to break down.
There are two forms of Pompe Disease: Infantile Onset Pompe Disease, or IOPD, in which symptoms occur in infancy, and Late Onset Pompe Disease, or LOPD, in which symptoms occur after infancy.
IOPD is more severe and progresses more rapidly than LOPD.
LOPD causes symptoms that may not be noticeable at first and can mimic other disorders. LOPD is difficult to diagnose and often misdiagnosed, in part because it’s a rare disease, but also because its symptoms can often feel disconnected and mistaken for other conditions. This can result in a long and frustrating diagnostic journey.
Fabry is a rare disease caused by changes in the GLA gene, which may be referred to as mutations or variants.
People with Fabry disease have trouble breaking down and getting rid of certain fatty waste substances (substrates) in cells. This happens because of a variant in the GLA gene that leads to a deficient or absent enzyme called alpha-Gal A. When alpha-Gal A is present and functioning, it travels to the cell’s recycling center, called the lysosome, to break down the fatty substances in the cell.
In Fabry disease, without fully functioning alpha-Gal A, the fatty substances build up in the body, causing damage to tissues and organs.
FSGS is a rare, serious kidney disorder characterized by progressive scarring (sclerosis) in parts of the glomeruli—the kidney’s filtering units. This scarring leads to proteinuria, progressive loss of kidney function, and often end-stage renal disease.
In the United States, more than 40,000 people are estimated to be living with FSGS, including both adults and children.
There are no therapies specifically approved for FSGS in the U.S. as of Nov. 2025.